Синдром Коффина-Лоури

Синдром Коффина-Лоури — это редкая генетическая болезнь с Х-сцепленным механизмом передачи, которая характеризуется выраженной умственной отсталостью, множественными фенотипическими особенностями. Пациенты имеют специфические черты внешности: высокий лоб, гипертелоризм, крупный выступающий нос. Помимо интеллектуальных нарушений, наблюдаются задержка роста, тугоухость, кардиологические осложнения. Основу диагностики синдрома Коффина-Лоури составляет молекулярно-генетическое исследование. Лечение поддерживающее: специальная программа нейрореабилитации, коррекция костных искривлений, слухопротезирование при тугоухости.
Общие сведения
Болезнь получила свое название в честь двух ученых, которые независимо друг от друга описали ее патогномоничные симптомы, — американского педиатра Грейнджа С. Коффина (1966 г.) и канадского генетика Роберта Б. Лоури (1971 г.). Молекулярно-генетические основы патологии были расшифрованы в 1996 году. Синдром Коффина-Лоури в полной клинической форме встречается только у больных мужского пола. Женщины, являющиеся носителями мутантного гена, могут иметь умеренные признаки синдрома. Заболевание регистрируется с частотой 1-2 случая на 100 000 живорожденных новорожденных.

Причины
Болезнь Коффина-Лоури связана с наличием мутации гена RPS6KA3 на Х-хромосоме. До 80% случаев патологии возникают спорадически в семьях без отягощенной наследственности, а оставшиеся 20% заболевших приходятся на Х-сцепленный рецессивный тип наследования. Клинические признаки проявляются преимущественно у мужчин. Факторы, способствующие спонтанным генетическим мутациям, пока не установлены.
Патогенез
В норме ген RPS6KA3 кодирует синтез белка киназы RSK2, необходимого для правильного протекания процессов пролиферации, дифференцировки, запрограммированной клеточной смерти. Вещество активизируется при помощи протеинов сигнального пути, начинает работать при поступлении гормонов роста, нейротрансмиттеров, полипептидных гормонов.
При дисфункции RPS6KA3 в организме наблюдается дефицит киназы, что становится причиной расстройств передачи по MARK-сигнальному пути. В результате этого ухудшается формирование долговременной памяти и других когнитивных функций, что обусловлено нарушениями синаптической передачи, а также возникают множественные патологии развития опорно-двигательного аппарата.
Симптомы
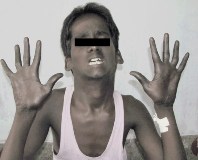
Наиболее характерные клинические проявления: многочисленные аномалии лицевого скелета. У детей с рождения высокий выступающий лоб, широко расставленные глаза с узкими глазными щелями. Переносица уплощенная, крылья носа крупные с вывернутыми ноздрями. Аномалии ротовой полости включают узкое высокое небо, изменения прикуса, срединную язычную борозду.
У страдающих синдромом Коффина-Лоури широкие кисти с короткими пальцами, которые резко сужаются к дистальным отделам. В 80% случаев возникает деформация грудной клетки, кифосколиоз. У мужчин отмечается серьезная задержка физического развития, средний рост взрослых составляет около 143 см (диапазон 115-158 см). Женщины-гетерозиготы зачастую имеют нормальную длину тела — более 50% пациенток находятся выше 10-го центиля.
С первых лет жизни у больных проявляется нарушение речевой функции, отставание в интеллектуальном развитии. Степень умственной отсталости широко варьирует, у разных пациентов уровень IQ может быть в диапазоне 15-60. При этом у ребенка частично сохранены коммуникативные навыки, характер обычно спокойный и дружелюбный. Тяжелые формы умственной отсталости, как правило, формируются при сопутствующей тугоухости.
Осложнения
До 20% больных страдают нарушениями координации движений, внезапными падениями (дроп-атаки), которые появляются в школьном возрасте. До 15% пациентов имеют сердечно-сосудистые осложнения (дисфункцию митрального клапана, кардиомиопатию). У 5% людей с синдромом Коффина-Лоури отмечаются эпилептические приступы. Гетерозиготные женщины часто имеют ожирение, психические проблемы: депрессии, шизофрению, психопатологический тип поведения.
Согласно литературным данным, около 13,5% мужчин погибают в 13-34 лет, остальные доживают до среднего возраста. Причинами летального исхода выступают негативные последствия синдрома Коффина-Лоури — миокардит, пневмония, дыхательная недостаточность. Также существует риск смерти от осложнений при проведении наркоза для ортопедической коррекции костных аномалий или выполнения стоматологических манипуляций.
Диагностика
Характерный фенотип в сочетании с нарушениями психомоторного развития позволяет заподозрить синдром Коффина-Лоури еще в младенчестве. Обследованием таких больных занимается врач-педиатр совместно с детским неврологом и генетиком. Чтобы подтвердить диагноз, необходима комплексная диагностика, включающая следующие методы:
- Генетическое тестирование. Секвенирование экзона для обнаружения типичной мутации RPS6KA3 — основной способ подтвердить наличие синдрома. Такая диагностика может проводиться другим членам семьи при подозрении на наследственный характер болезни.
- Рентгенография скелета. На снимках лицевого черепа обнаруживается утолщение костей, множественные деформации. Рентгенография позвоночника показывает кифосколиоз, узкие межпозвоночные промежутки. При исследовании конечностей выявляется укорочение трубчатых костей, широкие проксимальные и узкие дистальные фаланги пальцев.
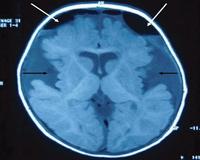
- Консультация невролога. Для оценки степени когнитивных и речевых расстройств требуется комплексный неврологический осмотр, в дошкольном и школьном возрасте применяются специальные тесты и опросники. По показаниям назначается нейровизуализация (КТ голоного мозга, нейросонография, ЭЭГ).
- Пренатальная диагностика. В семье с отягощенной наследственностью целесообразно исследовать генотип плода еще во время беременности. Забор материала для молекулярно-генетического теста выполняется с помощью амниоцентеза, кордоцентеза во втором триместре.
Лечение синдрома Коффина-Лоури
Консервативная терапия
Этиотропное лечение не разработано, поэтому медицинская помощь сводится к устранению или уменьшению симптоматики, предотвращению осложнений синдрома Коффина-Лоури. Чтобы контролировать состояние здоровья, пациентам необходимо динамическое наблюдение у ряда специалистов (педиатр/терапевт, невролог, кардиолог, генетик). Соответственно клинической картине назначаются:
- Психотропные препараты. Противосудорожные и ингибиторы обратного захвата серотонина рекомендованы при часто повторяющихся дроп-атаках. Регулярный прием антиконвульсантов показан, если заболевание осложняется судорожными приступами.
- Ноотропы. Использование лекарств, улучшающих энергообмен и кровообращение в головном мозге, стимулирует становление интеллектуальных функций, улучшает краткосрочную и долговременную память.
- Слухопротезирование. Детям с кондуктивной тугоухостью установка слухового аппарата выполняется как можно раньше, в идеале — в первые 6 месяцев жизни ребенка, что способствует развитию речи, правильному формированию коммуникативных навыков.
Учитывая умственную отсталость большинства больных, при синдроме Коффина-Лоури требуется специальная нейрореабилитация. Занятия начинаются с раннего возраста при участии логопедов, сурдопедагогов, коррекционных педагогов. Дети не могут обучаться в обычной школе, им требуются занятия в инклюзивных классах либо специальные программы домашнего обучения.
Хирургическое лечение
Оперативная ортопедическая помощь оказывается при 3-4 степени сколиоза, сопровождающегося кардиореспираторными нарушениями. Больным показана хирургическая коррекция искривления позвоночника с применением механической системы стабилизации
Прогноз и профилактика
Женщины-носители поврежденного гена имеют нормальную продолжительность жизни, симптомы выражены слабо или вовсе отсутствуют, поэтому прогноз благоприятный. Менее оптимистичны прогнозы для пациентов-мужчин, которые зачастую умирают в молодом и среднем возрасте от соматических или неврологических осложнений. Профилактика синдрома Коффина-Лоури включает медико-генетическое консультирование пар с семейной предрасположенностью.
|
Литература
1. Coffin—Lowry syndrome/ Marques Pereira, P., Schneider, A., Pannetier, S.// European Journal of Human Genetics. — 2010. — №18. 2. Altered neurodevelopment associated with mutations of RSK2: a morphometric MRI study of Coffin-Lowry syndrome/ Kesler S. R., Simensen R. J., Voeller K., Abidi F// Neurogenetics. — 2007. — №8. 3. Coffin-Lowry syndrome: clinical and molecular features/ Hanauer, A., and I. D. Young// Journal of medical genetics. — 2002. — №10. |
Код МКБ-10
Q87.0 |
| Процедуры и операции | Средняя цена |
| Психиатрия / Детская психиатрия и психология / Коррекция нарушений психической сферы у детей | 1938 р. 22 адреса |
| Отоларингология / Слухопротезирование | 2452 р. 22 адреса |
| Логопедия / Коррекция нарушений речи / Логопедическая помощь детям с особенностями развития | 1565 р. 12 адресов |
| Психиатрия / Психолого-психиатрическая помощь / Медикаментозное лечение нарушений психической сферы | 1992 р. 9 адресов |
| Логопедия / Коррекция нарушений речи / Логопедическая помощь детям с особенностями развития | 2175 р. 6 адресов |
| Логопедия / Коррекция нарушений речи / Логопедическая помощь детям с особенностями развития | 2500 р. 4 адреса |