Синдром Кронкхайта-Канада

Синдром Кронкхайта-Канада — редкий вариант диффузного полипоза органов ЖКТ, сочетающегося с лентигинозом, ониходистрофией, алопецией. Проявляется диареей, нелокализованными болями в брюшной полости, гиперпигментацией, выпадением волос, разрушением ногтей, атрофическим глосситом, ангулярным хейлитом, истощением. Диагностируется с помощью данных копрограммы, эзофагогастродуоденоскопии, колоноскопии, рентгенографии желудка, кишечника. Для лечения применяют кортикостероиды, производные кромоглициевой кислоты, блокаторы H2-гистаминорецепторов, антибиотики, ингибиторы протонной помпы. По показаниям проводят резекцию тонкого или толстого кишечника, стомирование.
Общие сведения
Первое описание синдрома было представлено в 1955 году американскими клиницистами Л.В. Кронкайтом и В.Дж. Канада. Заболеваемость не превышает 1 случая патологии на 1 млн. жителей. По данным исследований в сфере гастроэнтерологии и дерматологии, в период с 1955 по 2002 год выявлено 467 больных с расстройством Кронкхайта-Канада, 75,5% из которых проживало в Японии. Болезнь проявляется после 30 лет, средний возраст пациентов, по сообщениям разных авторов, составляет 55-59 лет. Мужчины заболевают в 1,3-1,5 раза чаще женщин. Актуальность своевременной диагностики синдрома связана с быстрым прогрессированием симптоматики, плохими показателями 5-летней выживаемости без адекватной терапии, высоким риском смертельных осложнений.

Причины синдрома Кронкхайта-Канада
Этиология заболевания не сегодняшний день не установлена. Ранее считалось, что расстройство наследуется по аутосомно-доминантному типу, однако убедительных доказательств семейной предрасположенности не получено. Кроме того, у больных отсутствуют мутации гена PTEN, характерные для других врожденных полипозов пищеварительного тракта (болезни Рувалкаба-Мири-Смита, синдрома Коудена и др.). В качестве одной из рабочих теорий происхождения патологии рассматривается развитие аутоиммунной реакции, что косвенно подтверждается выявлением в сыворотке антинуклеарных антител, гиперреактивностью тучных клеток, положительными результатами назначения кортикостероидов и мембраностабилизаторов.
Патогенез
Из-за неясности этиологии механизм развития заболевания изучен недостаточно. Пусковые моменты, провоцирующие возникновение диффузного полипоза, неизвестны. Патоморфологические изменения в слизистой кишечника проявляются расширением и кистозной трансформацией желез с образованием ретенционных кист, признаками поверхностного воспаления, резко выраженной отечностью стромы, умеренным утолщением и воспалительной инфильтрацией мышечного слоя. По мере прогрессирования патологии образовавшиеся полипы подвергаются множественным поверхностным некрозам. У большинства пациентов полипозные образования выявляются на слизистой желудка и толстого кишечника, реже поражается тонкий кишечник и пищевод.
Ведущую роль в патогенезе синдрома Кронкхайта-Канада играет расстройство секреторной, пищеварительной и всасывающей функций пораженных отделов ЖКТ в сочетании с нарушениями моторики кишечной стенки. Результатом хронической диареи и недостаточности пристеночного пищеварения становится интенсивная потеря белка и микроэлементов, приводящая к развитию гипопротеинемии, гипокалиемии, гипокальциемии, гипомагниемии с соответствующей клинической симптоматикой. Нарушения белково-минерального обмена, вероятнее всего, служат основной причиной кожных проявлений болезни.
Симптомы синдрома Кронкхайта-Канада
Клиническая картина заболевания нарастает постепенно. Первым признаком болезни становится диспепсический синдром, который проявляется обильным жидким стулом с примесями слизи до 5-7 раз в сутки, болезненностью в животе без четкой локализации. Спустя несколько месяцев после начала желудочно-кишечных расстройств возникают характерные кожные симптомы – у пациентов выявляются гиперпигментированные пятна до 10 см в диаметре, расположенные на кистях рук, лице, шее, бедрах, голенях, тыльной поверхности стоп, изредка на слизистых оболочках. У некоторых больных на коже появляется депигментация (витилиго).
Постоянным симптомом болезни Кронкхайта-Канада является истончение и ломкость ногтей, в тяжелых случаях происходит полное отслоение ногтевой пластины. На поздних стадиях синдрома присоединяются отеки (в основном на нижних конечностях), судороги, наблюдается диффузное выпадение волос. Характерны изменения со стороны слизистой ротовой полости: язык становится опухшим, болезненным, на нем образуются красные пятна, изъязвления, в уголках рта формируются пузырьки, эрозии, гнойно-кровянистые корки. Пациенты стремительно теряют массу тела вплоть до развития кахексии.
Осложнения
При симптомокомплексе Кронкхайта-Канада существует риск изъязвления полипов с профузными желудочно-кишечными кровотечениями. В случае малой хронической кровопотери возникает железодефицитная анемия с бледностью, снижением трудоспособности, головокружениями, гипотонией, тахикардией. Вследствие нарушения процессов переваривания и всасывания питательных веществ развивается синдром мальабсорбции, что приводит к гипопротеинемии, дистрофии мышечной ткани, в том числе миокардиодистрофии, остеомаляции, дефициту микроэлементов, значительному снижению иммунитета. При полипах большого размера может развиваться механическая кишечная непроходимость. У 13% пациентов полипы малигнизируются, в 8% случаев возникает аденокарцинома толстого кишечника, в 5% — желудка. Основными причинами смерти больных становятся кишечные кровотечения, сердечная недостаточность, сепсис.
Диагностика
Постановка диагноза зачастую затруднена, что обусловлено незначительной распространенностью заболевания и сходством клинической картины с другими болезнями желудочно-кишечного тракта. О возможном наличии синдрома Кронкхайта-Канада свидетельствует сочетание диспепсических проявлений с гиперпигментацией, ониходистрофией, алопецией. Наиболее информативны для постановки диагноза:
- Копрограмма. В лабораторном анализе кала определяются признаки синдрома мальабсорбции: большое количество нейтральных жиров, непереваренных мышечных волокон, зерен крахмала. Дополнительно для обнаружения скрытой крови в кале, свидетельствующей о хроническом кровотечении из ЖКТ), проводится реакция Грегерсена.
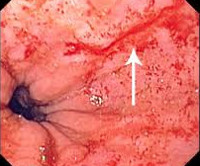
- Эзофагогастродуоденоскопия. При эндоскопическом исследовании верхних отделов пищеварительного тракта оценивается состояние слизистой оболочки, выявляются типичные полипозные разрастания эпителия. Для цитологического исследования тканей выполняется биопсия измененных участков.
- Колоноскопия. При наличии у пациента синдрома дистального колита целесообразно исследование толстого кишечника гибким эндоскопом. В ходе осмотра врач обнаруживает диффузный полипоз, отбирает биоптаты для гистологического анализа, по показаниям проводит лечебные мероприятия (остановку кровотечения, резекцию).
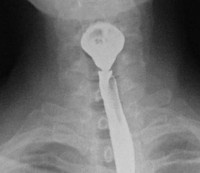
- Рентгенография желудка и кишечника. Серия рентгеновских снимков после перорального введения контрастного вещества при заболевании Кронкхайта-Канада позволяет визуализировать полипы в виде округлых дефектов наполнения с четкими контурами. Метод высокоинформативен при размере образований более 5 мм.
В общем анализе крови обнаруживают снижение уровня эритроцитов и гемоглобина, микроцитоз. В биохимическом анализе крови наблюдается гипопротеинемия, уменьшение концентрации электролитов (калия, магния, кальция). Проведение ангиографии кишечных сосудов позволяет дифференцировать полипы с интрамуральными опухолями. При недостаточной информативности других методик назначается КТ брюшной полости. Дифференциальная диагностика осуществляется с истинными и гиперпластическими полипами, синдромом Пейтца-Егерса, язвенной болезнью, опухолями пищеварительного тракта мезенхимального и эндотелиального происхождения, болезнью Менетрие, ювенильным полипозом, синдромом Гарднера, кишечной лимфангиэктазией. Кроме осмотра гастроэнтеролога пациенту рекомендованы консультации онколога, гематолога, генетика, дерматолога.
Лечение синдрома Кронкхайта-Канада
Выбор врачебной тактики зависит от локализации и масштаба распространения диффузного полипоза. При поражении желудка, тонкого кишечника, пищевода рекомендовано консервативное ведение пациента с назначением пробной противовоспалительной терапии и коррекцией возникших расстройств метаболизма. Диета аналогична рациону при других энтеропатиях: рекомендовано ограничение количества жиров (не более 30 г/сут), употребление продуктов, богатых белками, жирорастворимыми витаминами, основными макроэлементами, прием специальных жировых эмульсий, содержащих среднецепочечные триглицериды. При значительном снижении протеинов крови пациентам показано парентеральное питание с введением белково-содержащих растворов (2-3 одномесячных курса инфузий в год). Схема медикаментозного лечения включает:
- Кортикостероиды. На фоне гормональной терапии обычно уменьшается диарея, улучшаются процессы пищеварения. Отмечается стабилизация дерматологических симптомов: замедляется или прекращается выпадение волос, восстанавливается структура ногтей, цвет кожи становится более светлым и однородным.
- Стабилизаторы мембран тучных клеток. Улучшению состояния часто способствует дополнение гормонотерапии производными кромоглициевой кислоты. Угнетение кальций-зависимой дегрануляции тучных клеток тормозит выделение из них гистамина, предотвращает или уменьшает развитие местной воспалительной реакции.
- Н2-гистаминоблокаторы. При преимущественном поражении диффузным полипозом Кронкхайта-Канада слизистой желудка эффективен прием конкурентных ингибиторов гистаминовых рецепторов. Количество полипов заметно уменьшается на фоне снижения базальной и стимулированной секреции соляной кислоты.
По результатам наблюдений, динамика течения синдрома улучшается при назначении ингибиторов протонной помпы, макролидов, β-лактамных пенициллинов, других антибактериальных препаратов для элиминации хеликобактерий. Хирургические методы показаны при локальном поражении кишечника – проводится сегментарная, лапароскопическая резекция тонкой кишки, удаление различных отделов толстой кишки с выполнением при необходимости колостомии, сигмостомии, цекостомии, последующих реконструктивных вмешательств.
Прогноз и профилактика
Исход заболевания зависит от выраженности экссудативной энтеропатии и наличия осложнений со стороны других органов. Прогноз при расстройстве Кронкхайта-Канада неблагоприятный, около 45% пациентов умирают в течение первых 5 лет после манифестации клинических проявлений. Меры специфической профилактики не разработаны. Для предупреждения развития осложнений необходимо динамическое наблюдение за больными с установленным диагнозом диффузного полипозного синдрома, которое включает регулярное посещение гастроэнтеролога, ежегодное выполнение диагностического эндоскопического исследования.
|
Литература
1. Секреты гастроэнтерологии/ МакНелли П.Р. – 2005. 2. Полипы желудочно-кишечного тракта/ Юхтин В.И. – 1978. |
Код МКБ-10
D13 |
| Процедуры и операции | Средняя цена |
| Дерматология / Консультации в дерматологии | от 130 р. 1848 адресов |
| Гастроэнтерология / Консультации в гастроэнтерологии | от 37 р. 1646 адресов |
| Проктология / Консультации в проктологии | от 253 р. 1013 адресов |
| Онкология / Консультации в онкологии и гематологии | от 56 р. 927 адресов |
| Диагностика / Компьютерная томография (КТ) / КТ внутренних органов | от 600 р. 886 адресов |
| Проктология / Диагностика в проктологии / Эндоскопия в проктологии | от 250 р. 822 адреса |
| Гастроэнтерология / Диагностика в гастроэнтерологии / Рентгенография в гастроэнтерологии | от 185 р. 476 адресов |
| Онкология / Консультации в онкологии и гематологии | от 345 р. 401 адрес |
| Гастроэнтерология / Диагностика в гастроэнтерологии / Рентгенография в гастроэнтерологии | от 185 р. 265 адресов |
| Гастроэнтерология / Диагностика в гастроэнтерологии / Эндоскопия в гастроэнтерологии | от 350 р. 234 адреса |
Комментарии к статье
Вы можете поделиться своей историей болезни, что Вам помогло при лечении синдрома Кронкхайта-Канада.